En el marco de la actual incertidumbre económica y del proceso de deslocalización que vive la industria farmacéutica, la investigación clínica sigue siendo de las actividades en I+D+i que conducen a los países hacía la vanguardia y el progreso. Sin embargo, existe un gran número de dificultades que la industria farmacéutica en España debe solventar, con diligencia, si pretende seguir participando en ensayos clínicos y proyectos de investigación.
El futuro de nuestra Sanidad es cada vez más incierto, tanto por los recientes recortes en los precios de los medicamentos, como por la problemática de las farmacias para dispensarlos, debido al retraso de los pagos por parte de las administraciones.
Actualmente, el sector farmacéutico sigue siendo de los pocos que realmente apuesta por el I+D+i en España. La investigación clínica, por parte de la industria farmacéutica, permite determinar o confirmar los efectos clínicos, farmacológicos y/o demás efectos farmacodinámicos, y/o detectar las reacciones adversas, y/o estudiar la absorción, distribución, metabolismo y eliminación de uno o varios medicamentos en investigación, con el fin de determinar su seguridad y/o eficacia y, en último término, mejorar o restituir la salud de los pacientes. Aun así, a estos fármacos se les exige que sean cada vez más coste-efectivos para garantizar un espacio en las farmacias hospitalarias y en las oficinas de farmacia; y ello, dentro de un entorno tan cambiante y tan marcado por la crisis como el actual. Además, la realización de Ensayos Clínicos (EC) permite conocer de primera mano, a la comunidad médica, el manejo de nuevos medicamentos y conocer mejor el perfil adecuado de los pacientes.
La investigación clínica, tan larga y costosa, adquiere una complejidad adicional, al existir distintos niveles de toma de decisiones: Ministerio de Sanidad, CCAA y todas las Comisiones Hospitalarias de Farmacia y Terapéutica. Cada vez más, todas ellas requieren de nuestros fármacos más evidencia científica, y también fármacoeconómica. Sin embargo, existen muchos obstáculos que podrían evitarse si, previamente, las Autoridades Sanitarias se sentaran conjuntamente con la industria farmacéutica, en fases clínicas más tempranas, donde actualmente no existe un foro de consenso, con el fin de poder exponer las necesidades clínicas, y así, diseñar correctamente los EC, que son la única vía que permite aportar y garantizar la eficacia y seguridad de nuestros medicamentos. Por tanto, a mi modo de ver, deberían formularse conjuntamente una pregunta fundamental para el éxito de los futuros fármacos: ¿Qué requisitos debe cumplir el fármaco clínica y fármaco-económicamente para que, a posteriori, si todos éstos se cumplen, la aprobación sea directa y los pacientes puedan ser tratados lo antes posible?
En último término, toda esta complejidad redunda en una posible pérdida de competitividad de las filiales españolas, dentro de las multinacionales farmacéuticas, que podría ocasionar la disminución o desaparición de la participación en EC.
En base a mi experiencia personal de los 2 últimos años en Otsuka Pharmaceutical, sobre Estudios Posautorización1 (EPA), relacionados con la hiponatremia y el Síndrome de Secreción Inapropiada de Hormona Antidiurética (SIADH), me gustaría trasladar las siguientes consideraciones, de orden práctico, que pueden contribuir al éxito de un EC.
En primer lugar, sea cual sea la fase a la que pertenezca el estudio, es fundamental realizar un buen diseño del mismo, el cual será plasmado en el protocolo (asegurando el cumplimiento de las normas de Buena Práctica Clínica [BPC] y éticamente justificable cumpliendo con los principios éticos básicos contenidos en la Declaración de Helsinki de la Asociación Médica Mundial sobre principios éticos para las investigaciones médicas en seres humanos, y en sus posteriores revisiones). Por nuestra parte, sería deseable incluir la opinión de las Autoridades Sanitarias en el diseño de los EC, y diseñar estudios head-to-head, escogiendo, en la medida de lo posible, como elemento comparativo, el tratamiento gold standard; utilizar resultados clínicos relevantes y monitorizar la utilización de recursos sanitarios, con el fin de demostrar si es más eficaz, seguro a largo plazo y más coste-efectivo que las herramientas terapéuticas ya existentes.
En segundo lugar, la generación de un partnership real de la Contract Research Organization (CRO) y la industria farmacéutica es un elemento fundamental para llevar a cabo un EC. Por consiguiente, la clave está en encontrar aquella CRO que sea excelente y competente, y que nos aporte el know-how adecuado sobre los diferentes aspectos que le sean asignados (Dirección de Proyectos, Dirección Médica, Data Management y Bioestadística, Tecnologías de la Información y la Comunicación, Regulatorio/Legal y Financiero) para garantizar la optimización en el desarrollo del EC.
Este partnership se dificulta en el caso de estudios internacionales o promotor extranjero, y gestionados por una CRO internacional que, a su vez, tiene subcontratada a otra CRO nacional.
En tercer lugar, otro de los puntos clave sería entender el mecanismo de cada Comité Ético de Investigación Clínica (CEIC) de los hospitales donde se realice el EC, y sus plazos de resolución, en la notificación, enmiendas y emisión del dictamen. Si nos detenemos en este punto, destacar que sería ideal en la práctica, disponer de un sistema de CEIC de aprobación central, que habilitara simultáneamente a todos los hospitales.
Profundizando más, en el contexto de EC Fase IV (realizados con un medicamento después de su comercialización), se publicaron las Rutas Administrativas de los Estudios Posautorización (Figura 1) sobre los EPA de tipo Observacional con medicamentos de uso humano, según Directrices de la Orden SAS/3470/2009, los cuales ponen de manifiesto todos los pasos a realizar para poder iniciar un estudio.
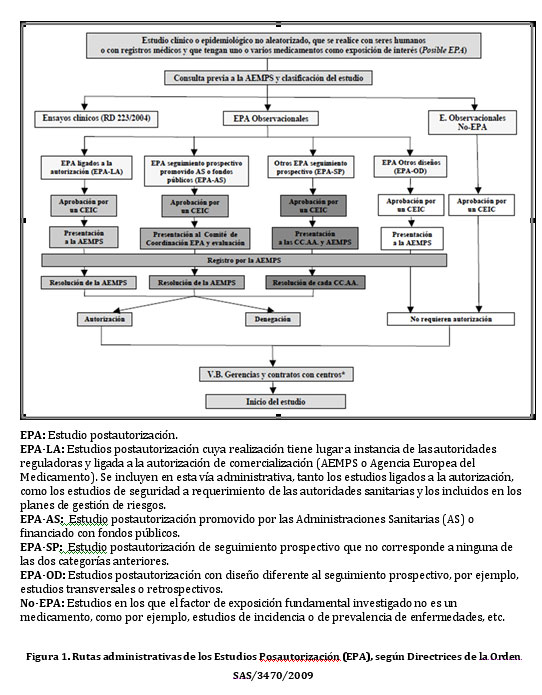
Con referencia al tema de los CEIC, realmente no quedó del todo claro si con el dictamen del CEIC del hospital principal acreditado era suficiente, o si era preciso evaluar adicionalmente por los otros CEIC. A posteriori, se publicó el pasado marzo de este año 2011, el Documento de Preguntas y Respuestas sobre Estudios Posautorización de tipo Observacional con Medicamentos de Uso Humano y sobre la Aplicación de la Orden SAS/3470/2009, con el fin de avanzar en la armonización de criterios para la realización de este tipos de estudios en el territorio nacional. Pero, aun así, a día de hoy en la práctica, quedan sin resolver incógnitas sobre los entresijos de los CEIC.
Y por último, y no menos importante, no olvidar que, en los EC Fase IV, antes de incluir a un centro hospitalario, previamente tenga aprobado el fármaco en su guía farmacoterapéutica.
En conclusión, en general, para una buena consecución de un EC es fundamental:
* Establecer un consenso previo con las Autoridades Sanitarias sobre los requerimientos clínicos y farmacoeconómicos, en el diseño de los EC, especialmente en fases tempranas de su desarrollo, para su autorización.
* Elección de una CRO excelente y competente, con la cual generar un estrecho partnership.
* Comprender el mecanismo de cada CEIC hospitalario.
* En EC Fase IV, tener aprobado previamente el medicamento en todas las guías farmacoterapéuticas de todos los centros participantes.
1 He estado involucrado en diversos EC, los cuales son: un Estudio Nacional transversal, observacional y multicéntrico, clasificado como EPA-OD (con diseño diferente al de seguimiento prospectivo, por ejemplo, estudios transversales o retrospectivos); un Registro Internacional (EU y EUA) observacional, prospectivo y multicéntrico, clasificado como EPA-SP (de seguimiento prospectivo); y un Estudio Europeo observacional de seguridad, posterior a la autorización (Post-Authorisation Safety Study; PASS) y multicéntrico, inducido por la EMA, clasificado como EPA-LA (ligado a autorización; estudios de seguridad).
Nota del editor: Las opiniones del autor son exclusivamente a título personal.











